segunda-feira, 24 de maio de 2010
domingo, 23 de maio de 2010
quem somos nós.
Mieloma Múltiplo
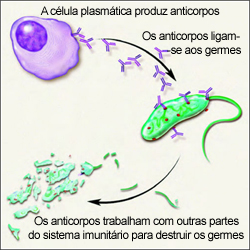
O mieloma múltiplo é um cancro que tem origem nas células plasmáticas, um tipo de glóbulos brancos.
O mieloma, à semelhança dos outros cancros, tem origem nas células, a unidade básica do organismo. Normalmente, as células crescem e dividem-se para formar novas células à medida que o organismo delas necessita. Quando as células envelhecem, morrem e são substituídas por novas células. No cancro, este processo ocorre de forma desordenada e as células continuam a dividir-se mesmo quando não são necessárias. Assim formam-se novas células sem que o organismo delas necessite, e sem que as células envelhecidas morram quando deviam. Este excesso de células pode dar origem à formação de uma massa de tecido a que se dá o nome de neoplasia ou tumor.
O mieloma desenvolve-se quando uma célula plasmática se torna anómala. As células anómalas dividem-se para formar cópias de si próprias. As novas células dividem-se uma e outra vez, produzindo mais e mais células anómalas. As células plasmáticas anómalas são as células do mieloma. Estas células produzem anticorpos designados por proteínas monoclonais (M).
Com o tempo, as células de mieloma acumulam-se na medula óssea impedindo o desenvolvimento normal das células sanguíneas. As células de mieloma também se acumulam na parte sólida do osso. A doença é designada por “mieloma múltiplo” uma vez que afecta múltiplos ossos. (Se as células do mieloma apenas se acumularem num osso, essa “massa única” recebe o nome de plasmacitoma.)
As células de mieloma (células plasmáticas anómalas) produzem proteínas monoclonais.
O mieloma múltiplo é o tipo mais comum de tumor das células plasmáticas. Nesta secção não são abordados outros tipos de tumores das células plasmáticas.
O mieloma múltiplo não é um carcinoma dos ossos.
Apesar de afectar os ossos, mieloma múltiplo tem origem nas células sanguíneas e não nas células do osso.
O cancro ósseo é uma doença diferente. Tem origem nas células ósseas e não nas células sanguíneas.
O cancro ósseo é diagnosticado e tratado de forma diferente do mieloma múltiplo.
Sintomas:
- Dor nos ossos
- Fracturas (ossos partidos) espontâneas
- Sentir-se fraco e muito cansado
- Infecções e febre frequente
- Perda de peso
- Náuseas ou prisão de ventre.
Factores de risco:
- Idade: O avançar da idade aumenta a probabilidade de desenvolver mieloma múltiplo. A maioria dos casos de mieloma é diagnosticada em pessoas com mais de 65 anos. Esta doença raramente ocorre antes dos 40 anos de idade.
- Etnia. O risco de mieloma múltiplo é mais elevado nos afro-americanos e mais baixo nos asiático-americanos. Desconhece-se a razão para esta diferença entre grupos étnicos.
- História de gamapatia monoclonal de significado indeterminado (GMSI): A GMSI é uma afecção em que as células plasmáticas anómalas produzem níveis baixos de proteínas M. A GMSI é uma doença benigna, mas aumenta o risco de desenvolver mieloma múltiplo.
Diagnóstico:
- Análises ao sangue: O laboratório determina o nível de células sanguíneas e de outras substâncias no sangue. O mieloma faz aumentar o nível de células plasmáticas e de cálcio. A maioria das pessoas com mieloma tem anemia. O mieloma também faz aumentar o nível de algumas proteínas, como a proteína M, a beta-2-microglobulina, entre outras.
- Análises à urina: O laboratório avalia e quantifica os níveis de proteína de Bence Jones, um tipo de proteína M, na urina durante 24 horas.Se os níveis desta proteína estiverem elevados, a sua função renal deverá ser vigiada, uma vez que esta pode obstruir e danificar os rins.
- Raios X: Pode realizar uma radiografia para detectar as lesões típicas do mieloma múltiplo, bem como a existência de fracturas.
- Biopsia óssea: O médico recolhe tecido para procurar células cancerígenas. A biopsia é a única forma segura de saber se existem células de mieloma na medula óssea. É colhida uma amostra de medula óssea a partir do osso ilíaco, mediante anestesia local, para diminuir o desconforto. O patologista determina a existência de células cancerígenas, através de observação microscópica da amostra de medula óssea.
- Aspiração da medula óssea: O médico utiliza uma agulha fina para recolher amostras de medula óssea.
- Biopsia óssea: O médico utiliza uma agulha grossa para recolher uma pequena quantidade de osso.
Tratamento:
A escolha do tratamento depende sobretudo do estádio da doença e dos sintomas que o doente apresenta. O estado de saúde do doente será avaliado regularmente para se dar início ao tratamento quando o médico julgar pertinente. Por vezes, o transplante de células estaminais ou radioterapia fazem parte do plano de tratamento. O médico pode descrever as opções de tratamento e os resultados esperados para cada uma delas. O doente e o médico podem trabalhar em conjunto na elaboração de um plano de tratamento adaptado às necessidades do doente.
Transplante de células estaminais:
Alguns doentes com mieloma múltiplo são submetidos a um transplante de células estaminais. O transplante de células estaminais obriga a que o doente receba doses elevadas de quimioterapia, radioterapia ou ambas. As doses elevadas destroem tanto as células do mieloma como as células sanguíneas normais na medula óssea. Depois disso, o doente recebe células estaminais saudáveis através de uma veia de grande calibre. A partir das células estaminais transplantadas irão desenvolver-se novas células sanguíneas.
- Transplante autólogo de células estaminais: Neste tipo de transplante utilizam-se as células estaminais do próprio doente após realização do tratamento para o mieloma. As células estaminais são retiradas do doente. Estas podem ser tratadas para matar as eventuais células de mieloma que possam estar presentes. As células estaminais são congeladas e armazenadas. Depois de o doente ter recebido doses elevadas de tratamento, as células estaminais armazenadas são reinfundidas no doente.
- Transplante alogénico de células estaminais: Neste caso, as células estaminais saudáveis são provenientes de um dador. O dador pode ser um familiar directo ou uma pessoa compatível, embora sem qualquer grau de parentesco. Para garantir que as células do dador são compatíveis com as do doente, realizam-se análises ao sangue.
- Transplante singénico de células estaminais: Este tipo de transplante utiliza células estaminais de um irmão gémeo (gémeo verdadeiro) do doente.
Melanoma

O melanoma é o tipo de cancro da pele mais grave. Em Portugal surgem, anualmente, cerca de 700 novos casos de melanoma maligno.
O melanoma é um tipo de cancro de pele. Tem início nas células da pele, os melanócitos. Para perceber o melanoma, é útil conhecer a pele e os melanócitos: qual a sua função, como crescem e o que acontece, quando se tornam cancerígenos. A pele é o maior órgão do corpo: protege-o do calor, da luz do sol, de feridas e de infecções. Ajuda a regular a temperatura corporal, armazena água e gordura, e produz vitamina D. A pele tem duas camadas principais: a epiderme (exterior) e a derme (interior). A derme contém vasos sanguíneos, vasos linfáticos, folículos pilosos e glândulas. Algumas destas glândulas produzem suor, que ajuda a regular a temperatura do organismo. Outras glândulas produzem sebo, uma substância oleosa que contribui para que a pele não seque. O suor e o sebo atingem a superfície da pele, através de pequenas aberturas: poros. O melanoma surge quando os melanócitos (células pigmentares) se tornam malignos. A maioria das células pigmentares encontra-se na pele; quando o melanoma tem início na pele, a doença chama-se melanoma cutâneo. O melanoma pode, também, ocorrer nos olhos (melanoma ocular ou melanoma intra-ocular). O melanoma raramente surge nas meninges, no aparelho digestivo, nos gânglios linfáticos ou noutras áreas onde há melanócitos. Os melanomas com origem noutras zonas, que não a pele, não serão aqui abordados. O melanoma é um dos tipos de cancro mais comum. A probabilidade de desenvolver melanoma aumenta com a idade, embora a doença afecte pessoas de todas as idades. Pode ocorrer em qualquer superfície da pele. Nos homens, o melanoma encontra-se, muitas vezes, no tronco (zona entre os ombros e as ancas), ou na cabeça e pescoço. Nas mulheres, desenvolve-se muitas vezes na zona inferior das pernas. A ocorrência de melanoma, na raça negra e noutras raças com pele escura, é rara; quando se desenvolve em pessoas de pele escura, tende a ocorrer sob as unhas dos pés e mãos, na palma das mãos ou planta dos pés. Quando o melanoma se espalha, ou dissemina, podem aparecer células cancerígenas nos gânglios linfáticos vizinhos. Os gânglios linfáticos "captam" bactérias, células cancerígenas ou outras substâncias nocivas, que possam estar presentes no sistema linfático. Se o tumor atingiu os gânglios linfáticos, pode significar que as células cancerígenas se espalharam já para outras partes do corpo, tal como o fígado, pulmões ou cérebro. Neste caso, as células cancerígenas do "novo tumor" são, ainda, células de melanoma, e a doença chama-se melanoma metastizado, e não cancro do fígado, do pulmão ou do cérebro (sistema nervoso central).
- Bordos: as margens são geralmente irregulares, biseladas, parecendo borradas ou irregulares; o pigmento pode espalhar-se para a pele circundante.
- Cor: a cor é desigual; pode apresentar sombras de preto, castanho e um tom bronzeado.
- Diâmetro: existe uma alteração no tamanho, que geralmente aumenta. Os melanomas são, por norma, maiores do que a borracha de um lápis (5 milímetros).
- Muitos sinais comuns (mais de 50): ter muitos sinais aumenta o risco de desenvolver melanoma.
- Pele clara: o melanoma ocorre com maior frequência em pessoas com pele clara, que queima e "faz" sardas facilmente (geralmente, estas pessoas têm cabelo ruivo ou louro e olhos azuis), comparativamente a pessoas com pele escura. As pessoas de raça caucasiana (branca) desenvolvem melanoma mais frequentemente do que as de raça negra, provavelmente porque a pele clara sofre mais facilmente os danos causados pelo sol.
- História pessoal de melanoma ou cancro da pele: as pessoas que já foram tratadas a um melanoma, apresentam risco mais elevado de ter um segundo melanoma. Algumas pessoas desenvolvem mais de dois melanomas. Pessoas que tiveram um ou mais cancros de pele comuns, como o carcinoma das células basais ou carcinoma das células escamosas, têm risco aumentado para melanoma.
- Sistema imunitário enfraquecido (deprimido): pessoas cujo sistema imunitário está enfraquecido por certos tumores, por fármacos administrados depois de um transplante de órgãos ou por HIV, têm risco aumentado de desenvolver melanoma.
- Queimaduras solares graves, com feridas ou bolhas: uma pessoa que tenha tido pelo menos uma queimadura solar grave, com formação de bolhas, quando criança ou adolescente, tem risco aumentado de melanoma. Como tal, é aconselhável que os pais protejam a pele das crianças do sol. A protecção solar pode reduzir o risco de melanoma, mais tarde na vida. As queimaduras solares, em idade adulta, também são um factor de risco para melanoma.
- Radiação UV (ultra-violeta): pensa-se que o aumento mundial do número de melanomas esteja relacionado com o aumento do tempo de exposição ao sol. O melanoma é mais comum em zonas com grande incidência de radiação UV do sol. A radiação solar UV provoca envelhecimento prematuro e danos na pele, que podem originar melanoma. As fontes artificiais de radiação UV, como lâmpadas solares e cabines de bronzeamento (solários), também podem provocar danos na pele e aumentar o risco de melanoma. A exposição aos raios UV naturais deve ser limitada; as fontes artificiais devem ser evitadas.
O médico pode aconselhar a consulta com um médico especialista. O melanoma pode ser tratado por diferentes especialistas, como sejam: dermatologista, cirurgião, oncologista, radioterapeuta e cirurgião plástico. Pode ter um especialista diferente, para cada tipo de tratamento que vá fazer.
Linfoma não-Hodgkin

O conhecimento acerca do Linfoma não Hodgkin tem vindo a aumentar, devido à investigação; continuam a ser estudadas as suas causas e a ser investigados novos processos de tratamento para esta doença. Graças à investigação contínua, pessoas com Linfoma não Hodgkin podem esperar uma melhor qualidade de vida, e menor probabilidade de morrer desta doença.
O Linfoma nâo Hodgkin é um tumor que tem início no sistema linfático.
O sistema linfático faz parte do sistema imunitário do nosso organismo. O sistema imunitário combate as infecções e outras doenças. No sistema linfático, uma rede de vasos linfáticos transporta um líquido transparente, chamado linfa. Os vasos linfáticos são "conduzidos" para os gânglios linfáticos (pequenos órgãos, de forma redonda). Nos gânglios linfáticos existem os linfócitos (um tipo de glóbulos brancos). Os gânglios linfáticos capturam e removem bactérias, e outras substâncias nocivas, que possam estar na linfa. Os gânglios linfáticos existem no pescoço, axilas, tórax, abdómen e virilhas. Outras partes do sistema linfático incluem as amígdalas, o baço e o timo. O tecido linfático também se encontra noutras partes do corpo, como o estômago, a pele e o intestino delgado. Existem muitos tipos de Linfoma não Hodgkin. Todos os tipos de linfoma têm início nas células do sistema linfático. O Linfoma não Hodgkin tem início quando um linfócito, uma célula B ou uma célula T se torna anómala. Geralmente, o Linfoma não Hodgkin começa numa célula B, num gânglio linfático. A célula anómala divide-se, para fazer cópias de si própria; as novas células dividem-se novamente, e novamente, originando mais e mais células anómalas. As células anómalas são células cancerígenas, que não morrem quando deviam. Não protegem o organismo de infecções nem de outras doenças. As células cancerígenas podem espalhar-se para quase todas as partes do organismo, ou seja, metastizar.
Tipos de Linfoma:
Quando o linfoma é detectado, o patologista reporta o tipo de linfoma. Os tipos mais comuns são o linfoma difuso de grandes células B, e o linfoma folicular.
Os linfomas podem ser agrupados pela sua velocidade de crescimento:
- Indolentes (também chamados de baixo-grau): crescem lentamente. Tendem a causar poucos sintomas.
- Agressivos (também chamados de grau intermédio ou de alto grau): crescem e espalham-se mais rapidamente. Tendem a causar sintomas graves. Com o tempo, muitos linfomas indolentes tornam-se linfomas agressivos.
Sintomas:
- Gânglios linfáticos inchados e indolores, no pescoço, axilas e virilhas.
- Perda inexplicável de peso.
- Febre, sem foco explicável.
- Suores nocturnos abundantes.
- Tosse, dificuldade respiratória ou dor no peito.
- Fraqueza e cansaço que não desaparecem.
- Dor, inchaço ou sensação de enfartamento no abdómen
Factores de risco:
- Sistema imunitário debilitado: ter um sistema imunitário debilitado, devido a um problema hereditário, a infecção por HIV (vírus da imunodeficiência humana), devido a certos fármacos ou outros factores, aumenta o risco de desenvolver Linfoma não Hodgkin.
- Determinadas infecções: ter certo tipo de infecções, pode aumentar o risco de desenvolver Linfoma não Hodgkin. No entanto, o linfoma não é contagioso; não se "apanha" linfoma de outra pessoa. Os principais tipos de infecção, que podem aumentar o risco de linfoma, são:
Vírus da imunodeficiência humana (HIV): o HIV é o vírus que provoca a SIDA (síndrome da imunodeficiência adquirida). As pessoas que estão infectadas com o HIV , têm maior risco de desenvolver certos tipos de Linfoma não Hodgkin.
-Vírus de Epstein-Barr (EBV): a infecção com EBV tem sido associada a um risco aumentado de linfoma.
- Helicobacter pylori : esta bactéria pode causar úlceras no estômago; pode, ainda, aumentar o risco da pessoa ter linfoma, no revestimento do estômago.
Vírus dos linfomas T humanos (HTLV-1): a infecção por HTLV -1 aumenta o risco de desenvolver linfoma e leucemia.
- Vírus da hepatite C: alguns estudos demonstraram um risco aumentado de linfoma, em pessoas com o vírus da hepatite C. É necessário continuar a investigar qual o papel do vírus da hepatite C.
- Idade: apesar do Linfoma não Hodgkin poder aparecer em pessoas jovens, a probabilidade de desenvolver esta doença aumenta com a idade. A maioria das pessoa com Linfoma não Hodgkin tem mais de 60 anos.
Prevenção:
- Análises sanguíneas: o laboratório faz a contagem de células do sangue.
- Radiografia ao tórax: importante para despistar sinais de doença no tórax; permite uma avaliação indirecta dos pulmões e do coração.
Tratamento:
A escolha do tratamento depende de vários factores, incluindo:
- Tipo de Linfoma não Hodgkin (EX. linfoma folicular).
- Estadio do linfoma.
- Velocidade de crescimento do linfoma, ou seja, se é um linfoma indolente ou agressivo.
- Idade.
- Outros problemas de saúde.
Os tipos de tratamento que existem para combater o Linfoma não-Hodgkin são a quimioterapia, a radioterapia e a Imunoterapia.
Linfoma de Hodgkin

Na doença de Hodgkin, as células do sistema linfático tornam-se anómalas e começam a dividir-se e a crescer a um ritmo demasiado rápido e de forma descontrolada e desordenada. Devido às suas características, o linfoma de Hodgkin é considerado uma neoplasia.
Dado que o tecido linfático está presente em diversas regiões do organismo, a doença de Hodgkin pode ter origem em praticamente qualquer parte. Esta doença pode ocorrer num único nódulo linfático, num grupo de nódulos linfáticos ou ainda noutras partes do sistema linfático, como a medula óssea ou o baço. Este tipo de cancro quando se dissemina, fá-lo de forma relativamente ordenada: de um grupo de nódulos linfáticos para outro. Por exemplo, se a doença de Hodgkin surgir em nódulos linfáticos do pescoço alastra primeiro para os nódulos situados acima das clavículas e só depois para os nódulos linfáticos debaixo dos braços e no tórax. Por fim, pode disseminar-se para qualquer outra parte do organismo.
Sintomas:
- Inchaço indolor nos nódulos linfáticos do pescoço, axila ou virilha
- Febre recorrente inexplicável
- Suores nocturnos
- Perda de peso inexplicável
- Comichão.
Factores de risco:
Desconhecem-se as causas da doença de Hodgkin e os médicos raramente conseguem explicar por que é que umas pessoas são afectadas e outras não. Existem, porém, algumas certezas: a doença de Hodgkin não é contagiosa, ou seja, não se "apanha" esta doença de outra pessoa.
- Idade/sexo - A doença de Hodgkin ocorre com maior frequência em pessoas entre os 15 e os 34 anos e depois dos 55 anos. É mais comum nos homens do que nas mulheres.
- Antecedentes familiares - Os irmãos e irmãs dos doentes com linfoma de Hodgkin têm uma probabilidade superior à média de vir a desenvolver a doença.
- Vírus - O vírus de Epstein-Barr é um agente infeccioso que pode estar associado a uma probabilidade acrescida de desenvolver a doença de Hodgkin.
Diagnóstico:
- Raio-X: A maioria das radiografias não requer qualquer preparação e demora apenas alguns minutos . A passagem de radiação através do organismo permite obter uma imagem. Esta imagem mostra os ossos, mas também dá indicações sobre os tecidos moles. Por exemplo, uma radiografia ao tórax pode revelar se o linfoma de Hodgkin invadiu os pulmões ou os gânglios linfáticos no tórax.
- TAC: utiliza raios X e produz fotografias semelhantes às radiografias. Todavia, as imagens são tiradas em diversas 'camadas', adquirindo um aspecto tridimensional. Tal como a radiografia, pode revelar se o linfoma de Hodgkin invadiu os gânglios linfáticos e outros órgãos. Não é um procedimento doloroso ou difícil, embora demore mais tempo do que a radiografia (cerca de meia hora).Para a realização de algumas TACs não se pode comer ou beber durante algumas horas e implicam a injecção de uma substância de contraste. Para a realização de uma TAC a pessoa tem de passar através de um scanner e permanecer deitada durante todo o procedimento.
- RM (ressonância magnética): tal como a TAC, produz imagens tiradas em diferentes 'camadas', mas utiliza campos magnéticos para as produzirA ressonância magnética pode demorar cerca de uma hora durante a qual a pessoa tem de permanecer deitada e imóvel. Trata-se de um exame indolor, embora a máquina seja ruidosa. Uma vez que o scanner é constituído por um íman muito potente, devem ser retirados todos os objectos de metal (como jóias e relógios). As pessoas com implantes de metal (tais como monitores cardíacos, próteses articulares, placas, ou clips cirúrgicos) não devem realizar este exame. Algumas pessoas sentem medo de se sentirem fechadas durante o exame, pelo que será útil mencionar este facto antes do procedimento para que possa ser prestado auxílio adicional.
- Tomografia com Emissão de Positrões (PET)A PET, ou tomografia com emissão de positrões, é uma técnica altamente sensível que utiliza raios X para detectar partículas a partir de substâncias que foram injectadas no organismo. Isto permite aos médicos distinguir células 'activas' de linfoma, que podem causar a doença, e aglomerados de células inactivas. Esta técnica é de grande utilidade após o tratamento do linfoma de Hodgkin para se avaliar o grau de sucesso. Uma vez que se trata de equipamento dispendioso, a PET nem sempre está disponível.
Tratamento:
- Quimioterapia
- Radioterapia
- Transplantação
O médico elabora um plano de tratamento adaptado a cada doente. O tratamento da doença de Hodgkin depende do estádio em que se encontra a doença, da dimensão dos nódulos linfáticos dilatados, dos sintomas manifestados, da idade e do estado geral do doente, assim como de outros factores (o tratamento de crianças com doença de Hodgkin não é abordado nesta área).